

Research Article: Journal of Drug and Alcohol Research (2023) Volume 0, Issue 0
In Silico Molecular Docking and Admet Evaluation for Investigating the Anti-Cancer Potential of Ketoprofen Amides Against Cyclooxygenase-2 (Cox-2)
Muhammad Zeeshan1*, Sidra Batool1, Muhammad Usman Alvi1, Zarif Gul1, Maryam Ahmed1, Urooj Shahzadi2 and Hassan Abbas32Department of Chemistry, University of Agriculture, Faisalabad, Pakistan
3Department of Poultry Production, University of Veterinary and Animal Sciences, Lahore, Pakistan
Muhammad Zeeshan, Department of Chemistry, University of Okara, Okara, Pakistan, Email: m.zee1882@gmail.com
Received: 02-Jan-2023, Manuscript No. JDAR-23-88683; Editor assigned: 04-Jan-2023, Pre QC No. JDAR-23-88683 (PQ); Reviewed: 18-Jan-2023, QC No. JDAR-23-88683; Revised: 23-Jan-2023, Manuscript No. JDAR-23-88683 (R); Published: 30-Jan-2023, DOI: 10.4303/JDAR/236218
Abstract
Many epithelial cancers overexpress the inducible cyclooxygenase-2 COX-2, which is thought to be the cause of NSAID’s antitumor effects. While ketoprofens (Ket) are well-known NSAIDs and analgesics, their anticancer potential has not yet gotten much attention, despite the fact that they are used to treat mild to moderate pain, fever, and inflammation. Effective drugs can be expensive, time-consuming, and labor intensive to introduce in a traditional or customary method. Contrarily, computer-aided drug design, commonly referred to as In Silico drug design, is a relatively recent method for high-throughput screening of a vast database of chemicals. By producing hits for lead compounds in less time and at a lower cost, the In Silico virtual screening technique assists in the development of innovative medications. The compound 1 shows the least binding energy value which -9.9 while compound 2 shows the highest which is -7.7. The overall purpose of this study is to assess the potential of different ketoprofen amides and to determine the effectiveness of these amides on the target protein COX-2 (PDB ID: 3Q7D). Additionally, this research compares and characterizes the ADMET profiles of the target ligands, ketoprofen amides. The drug score revealed that ketoprofens adhere to all regulations, is not hazardous, and is not carcinogenic.
Keywords
NSAIDs; Ketoprofen amides; Cyclooxygenase (COX-2); Inhibitors; Molecular docking; ADMET
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs), according to several experimental, epidemiological, and clinical research, are potential anticancer drugs and may lower the chance of developing colon, lung, liver, and other cancers [1,2]. Many epithelial cancers overexpress the inducible cyclooxygenase-2 COX-2, which is thought to be the cause of NSAID’s antitumor effects, albeit the exact mechanism is yet uncertain [3]. The stimulation of apoptosis, blockage of angiogenesis, or direct reduction of cancer cell development by disrupting signal transduction pathways important for cell proliferation are further examples of the antineoplastic actions of NSAIDs [4]. Aspirin, sulindac, piroxicam, ibuprofen, and indomethacin are examples of non-selective COX-1/2 inhibitors. Celecoxib and NS 398 are examples of COX-2 selective inhibitors. Regardless of COX-2 expression, both have been demonstrated to have potent anti- proliferative effects, notably causing G1 cell cycle arrest or death, in a number of tumour cell lines. Together, our results imply that NSAIDs may at least partially exert their growth-inhibitory effects via COX-independent pathways. Cyclooxygenase-2 (COX-2) oxygenates arachidonic acid, 2-arachidonoylglycerol, and the endo-cannabinoids arachidonoylethanolamide [5]. Numerous mild, competitive inhibitors of arachidonic acid oxygenation were shown to be powerful inhibitors of endocannabinoid oxygenation when a series of COX-2 inhibitors (ketoprofenamides) were evaluated.
While ketoprofens (Ket) are well-known NSAIDs and analgesics, their anticancer potential has not yet gotten much attention, despite the fact that they are used to treat mild to moderate pain, fever, and inflammation [6]. Due to the relatively short plasma half-lives of many ketoprofen drugs, frequent dosages are necessary to sustain the therapeutic impact [7]. Numerous NSAID derivates have been created, serving as possible pro-drugs, to reduce adverse effects, lengthen plasma half-life, and boost water solubility or lipophilicity. For instance, many NSAID derivatives were produced as possible pro-drugs, including aliphatic and aromatic esters and amides, as well as amide derivatives with covalently attached anti-oxidant moieties [8]. The NSAID naproxen’s phenolic ester and amide derivatives have also been demonstrated to have anti-oxidative and anti-proliferative properties. In addition, the amide derivatives tended to be more potent cell proliferation inhibitors than similar esters, and they were all more potent than naproxen alone. [9].
Effective drugs can be expensive, time-consuming, and labor-intensive to introduce in a traditional or customary method [10]. For instance, high-throughput screening (HTS) uses multiple-well microplates and automated processing to assess a huge number of potential drug-like compounds to enhance drug development [11]. HTS should also have enough resources because processing specific HTS program costs money and necessitates the employment of robotic machines [12]. Contrarily, computer-aided drug design, commonly referred to as In Silico drug design, is a relatively recent method for high-throughput screening of a vast database of chemicals. By producing hits for lead compounds in less time and at a lower cost, the In Silico virtual screening technique assists in the development of innovative medications [13]. Improved In Silico drug design thus shortens the time needed to create, design, and optimize a new medicine. For years, the best lead compounds with diverse structural features for utilization with a certain biological target have been identified using the virtual screening method [14]. Furthermore, computer- aided drug design has been used to find a wide range of exciting medical applications and hits utilising virtual screening, molecular docking, and dynamics modelling techniques [15]. The overall purpose of this study is to assess the potential of different ketoprofen amides and to determine the effectiveness of these amides on the target protein COX-2 (PDB ID: 3Q7D). According to the study, there may be new ways to demonstrate how effective ketoprofen amides are in blocking the catalytic sites of the aforementioned proteins in the treatment of cancer. Additionally, this research compares and characterizes the ADMET (adsorption, distribution, metabolism, excretion, and toxicity) profiles of the target ligands, ketoprofen amides. The PyRx virtual screening tool’s default setup parameters were used for docking goals, and the greatest binding energy (kcal/ mol) with the negative sign was selected for more research. The binding relationship of the protein-ligand complex was then observed using the BIOVIA Discovery Studio Visualizer 2021. A web program called SwissADME was used to assess the drug-likeness and ADMET profiles (http://www. swissadme.ch) [16].
Methods
Data set
Ten compounds made up the dataset needed to perform this study. These compounds have been claimed to inhibit the COX-2 protein’s ability to cause cancer. The 2D and 3D optimized structures of the ligands used in the research are shown in Table 1.
| Compound | Name | Mol. Wt. | 2D Structure | 3D Optimized Structure |
|---|---|---|---|---|
| 1 | 2-(3-benzoylphenyl)-N-cyclohexylpropanamide | 335.44 |  |
 |
| 2 | 2-(3-benzoylphenyl)-N-(3-hydroxypropyl)propanamide | 311.37 |  |
 |
| 3 | 3-(2-(3-benzoylphenyl)propanamido)propanoic acid | 325.36 | 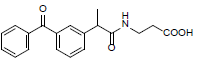 |
 |
| 4 | 2-(3-benzoylphenyl)-N-propylpropanamide | 295.38 | 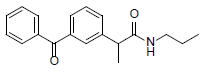 |
 |
| 5 | 2-(3-benzoylphenyl)-N,N-diethylpropanamide | 309.40 |  |
 |
| 6 | 2-(3-benzoylphenyl)-N-benzylpropanamide | 343.42 |  |
 |
| 7 | 2-(3-benzoylphenyl)-N-cyclohexylpropanamide | 335.44 |  |
 |
| 8 | 2-(3-benzoylphenyl)-N-phenethylpropanamide | 357.44 |  |
 |
| 9 | 2-(3-benzoylphenyl)-N-(2-hydroxyethyl)propanamide | 297.35 |  |
 |
| 10 | 2-(2-(3-benzoylphenyl)propanamido)acetic acid | 311.33 |  |
 |
Table 1: Information about ligands.
Protein preparation
The most appropriate 3D we used was (R)-naproxen bound to mCOX-2 [17] (PDB ID: 3Q7D) retrieved from the PDB database (www. pdb.org), as the protein target selected in our study was cyclooxygenase isoenzyme COX- 2. This structure has an R-value of 0.233 and a resolution of 2.340A. A pocket that was thought to be the active site was linked to each control. The residues at the active site were acquired using Biovia Discovery Studio 2021 [18]. To guarantee optimal binding interactions between the molecular targets and the ligands, water molecules, ligand groups, and any other sort of heteroatom on the proteins were removed and saved in PDB format. After that, PyRx was used to convert the proteins into macromolecules.
Ligand preparation
The ligands (1-10) were visualized and their geometry optimization was investigated at the B3LYP/6-31G (d,p) level of theory using Gaussian16 Rev. B.01 software [19]. After geometry optimization, three-dimensional structures were obtained in log format, which was then converted into PDB format using the Open Babel GUI [20] which the PyRx programme detected. Using PyRx software, the ligands were brought into their best conformations at the minimized energy, and they were then converted into the Auto-dock ligand format (pdbqt).
Molecular docking
To determine the binding affinities and identify probable binding sites, the molecular docking interactions between the proteins and the ligands were computed using the Autodock Vina of PyRx virtual screening programme [21]. A stochastic gradient optimization approach is used by the Vina wizard to forecast the binding affinities between ligands and molecular targets. Nine technical runs were used to dock in each case. Using Biovia Discovery Studio Visualizer 2021, the docking output interaction types with the greatest binding affinities, including hydrogen bonds and hydrophobic interactions, were visualized.
Drug-likeness evaluation and In Silico ADMET prediction
An online version of the SwissADME (http://www.swissadme. ch) web application was used to examine drug likenesses and the ADMET profiles [16]. For this investigation, all of the ligand’s Simplified Molecular Input Line Entry System (SMILES) forms were obtained, and the structures were sketched in ChemDraw. To determine whether each ligand’s drug-likeness falls within the acceptable range, Lipinski’s rule of five was utilized. The partition coefficient’s atom-based logarithm was used to examine the lipophilicity levels (ALogP). Human intestinal absorption, permeability (P)-glycoprotein inhibitor/substrate, and immortalized human colorectal adenocarcinoma cell line (Caco-2) data were examined to study the absorption of substances (ligands). The blood-brain barrier (BBB) was examined in relation to drug delivery. The CYP models (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4) for substrate or inhibitor were used to estimate drug metabolism. In addition to this, drug toxicity was examined, with a focus on hepatotoxicity, AMES toxicity, and suppression of the human ether-a-go-go-related gene (hERG). In order to identify Ketoprofen as a potential therapeutic candidate, all the relevant ADMET parameters of the compound were thoroughly estimated and confirmed for compliance with their standard ranges. Additionally, they were contrasted with the crucial parameters connected to other clinical medications used in the study in order to measure the potential of ketoprofens.
Results and Discussion
The goal of the study was to forecast the affinities of 10 ligands for the COX-2 target protein as hese compounds were reported to have anti-cancer activities [9]. The more relevant and practical the binding energy would be, the lower its value. The ADME properties of chemical compounds play a critical role in determining how well a medicine works. The difficult and demanding process of drug design and trial can be used to optimize ADME features, which can prevent pharmacokinetics-related failure in clinical stages [22]. Early consideration of ADME during the clinical drug development process has been shown to reduce attrition rates. As a consequence, 10 drug’s early-stage ADME features were evaluated using the SwissADME online tool.
Molecular docking analysis
The molecular docking simulation was used to verify the ketoprofen amide’s anticancer properties. The cyclooxygenase isoenzyme target COX-2 (PDB ID: 3Q7D) was docked against ten ligands in order to find potential binding interactions between the ketoprofen amides and the protein. Nine poses were acquired for all docking simulations and were assessed. Ketoprofen amides were successfully docked to the 3Q7D, and modes of interactions with a particular docking score were developed. The inhibitory effects of 10 ligand molecules on cyclooxygenase-2 COX-2 (3Q7D) were assessed in this study. While going through docking, protein (3Q7D) and drug molecules (ligands) do various things based on H-bond, hydrophobic interactions, Van der Waal interactions, and ionic bonds, etc., with their greatest docking score being the most stable for the ligand (Table 2). The RMSD and binding energy measurements are not necessarily the only criteria in molecular docking. Molecular interactions such as ionic bonds, hydrogen bonds, hydrophobic bonds, Van der Waal interactions, and others are also crucial.
| Protein | Ligand | Binding affinity | H-bond forming residues | Bond distance |
|---|---|---|---|---|
| (Kcal/mol) | (Å) | |||
| Cyclooxygenase-2 COX-2 (3Q7D) | 1 | -9.932 | Cys-41 | 1.93 |
| Cys-47 | 2.88 | |||
| Asn-39 | 2.62 | |||
| 2 | -7.745 | Gln-461 | 2.26 | |
| Asn-39 | 2.05 | |||
| 3 | -8.504 | Arg-120 | 2.78 | |
| 4 | -8.283 | Arg-120 | 2.01 | |
| 5 | -7.917 | Arg-120 | 2.8 | |
| 6 | -9.624 | Arg-376 | 2.39 | |
| Gln-374 | 2.73 | |||
| Arg-376 | 2.63 | |||
| 7 | -8.293 | Tyr-115 | 2.16 | |
| 8 | -8.88 | Arg-376 | 2.44 | |
| 9 | -7.889 | Glu-524 | 2.42 | |
| 10 | -8.641 | Arg-120 | 2.69 | |
| Glu-524 | 2.37 |
Table 2: Docking scores and the H-bond forming residues (with bond distance) results obtained between the interactions of ligands (1-10) and protein (3Q7D).
The linkage of certain amino acids involved in the interactions between drugs and proteins was also noted. In the active site of 3Q7D, Compound 1 (2-(3-benzoylphenyl)- N-cyclohexylpropanamide) formed H-bonds with Cys-41, Cys-47, and Asn-39, providing a binding affinity of -9.932 kcal/mol. Compound 1 showed some amide-pi stacked interaction with Val-155 (Figure 1).

Figure 1: Compound 1 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 1. (B) A docking model of interactions between compound 1 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
Compound 2 (2-(3-benzoylphenyl)-N-(3-hydroxypropyl) propanamide) has a binding energy of -7.745 kcal/mol while forming conventional H-bonds with Gln-461 and Asn-39 in the active site of 3Q7D (Figure 2). Compound 2 also formed some pi-alkyl interaction with Arg-44, Arg- 469, and Pro-153 along with 1 carbon-hydrogen bond with Arg-469.

Figure 2: Compound 2 docked in cyclooxygenase-2 COX-2 (3Q7D): (left) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 2. (right) A docking model of interactions between compound 2 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
Compound 3 (3-(2-(3-benzoylphenyl)propanamido)propanoic acid), showed significant binding, yielding a binding affinity of -8.504 kcal/mol while forming H-bonds with Arg-120 in the active site of 3Q7D. Some amide-pi stacked interaction of compound 3 with Tyr-115, pi-cation with Arg-120, pi-sigma with Val-89 and Val-116, and pi-alkyl with Leu-93 were observed (Figure 3).

Figure 3: Compound 3 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 3. (B) A docking model of interactions between compound 3 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
Significant binding was achieved by compound 4 (2-(3-benzoylphenyl)- N-propylpropanamide), which formed H-bonds with Arg-120 in the active site of 3Q7D and produced a binding affinity of -8.283 kcal/mol. Compound 4 was shown to have some interactions like pi-alkyl with Tyr- 355 and Leu-531, alkyl with Leu-93 and Val-349, pi-sigma with Ala-527 and Val-349, pi-pi T-shaped with Tyr-385 and Trp-387 and pi-sulfur with Met-522 (Figure 4).

Figure 4: Compound 4 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 4. (B) A docking model of interactions between compound 4 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
Compound 5 (2-(3-benzoylphenyl)-N,N-diethylpropanamide) interacted with 3Q7D with the binding affinity of -7.917 by forming hydrogen bonds with Arg-120 and also formed pi-cation with Arg-120, pi-alkyl with Val-116, pi-sigma with Val-89 and Leu-93 and alkyl with Pro-84, Val-89, and Ile-92 (Figure 5).

Figure 5: Compound 5 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 5. (B) A docking model of interactions between compound 5 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
By forming H-bonds with Arg-120 and Gln-374, as well as pi-sigma interactions with Leu-145, compound 6 (2-(3-benzoylphenyl)-N-benzylpropanamide), interacted with the 3Q7D with the binding affinity of -9.624 kcal/mol (Figure 6).

Figure 6: Compound 6 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 6. (B) A docking model of interactions between compound 6 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
Compound 7 (2-(3-benzoylphenyl)-N-cyclohexylpropanamide) bound to 3Q7D with the binding affinity of -8.293 kcal/mol by forming H-bonds with Tyr-115. Besides such strong hydrogen bonding, compound 7 formed pi-sigma interactions with Val-89, Val-116, and Leu-93. Some interaction of compound 7 like pi-alkyl with Leu-93 and Ile-92 and alkyl with Val-89 and Pro-84 were observed (Figure 7).

Figure 7: Compound 7 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 7. (B) A docking model of interactions between compound 7 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
Compound 8 formed H-bonds with Arg-376 in order to interact with 3Q7D with the binding affinity of -8.88 kcal/ mol. Compound 8 also generated pi-pi stacked contacts with Phe-142 in addition to such robust hydrogen bonds. There were some pi-sigma interactions between compound 8 and Leu-145 (Figure 8).

Figure 8: Compound 8 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 8. (B) A docking model of interactions between compound 8 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
Favorable hydrogen bond interactions between Glu-524 residues in the active site of 3Q7D were seen with compound 9 at the binding affinity of -7.889 kcal/mol. With Val-89, and Leu-93 residues, pi-sigma interactions were found. With Ile-112, Val-116, Leu-93, and Val-89 residues, the pi-alkyl bond type was also seen (Figure 9).

Figure 9: Compound 9 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 9. (B) A docking model of interactions between compound 9 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
The interactions of the compound 10 (2-(2-(3-benzoylphenyl) propanamido)acetic acid) to the 3Q7D with the binding affinity of -8.641 kcal/mol revealed that residues Arg- 120 and Glu-524 showed H-bonds. The residues Val-116, Leu-93, and Val-89 interacted with compound 10 through pi-sigma interaction while Ile-92 and Leu-93 showed pi-alkyl interaction (Figure 10).

Figure 10: Compound 10 docked in cyclooxygenase-2 COX-2 (3Q7D): (A) The H-bond donor-acceptor residues are visible in the pocket view of 3Q7D binding to compound 10. (B) A docking model of interactions between compound 10 and an amino acid with H-bonds in 3Q7D is shown in a 2D schematic picture.
ADME and toxicity results
Drug-likeness prediction: The ligand’s aqueous solubility and gut blood barrier permeability, which are connected to their drug-like characteristics, dictate the initial stage of oral bioavailability. Table 1 lists the findings for drug-likeness, bioactivities, ADME, and toxicity profile in the first stage of oral bioavailability. The evaluation of the compounds using Lipinski’s Rule of Five. These criteria, which consider molecular weight (Mol wt<500), are used to determine how frequently Lipinski’s rule of five has been violated. The topological surface area (TPSA<402Å2), number of hydrogen bond donors (nHBD<5) and acceptors (nHBA<10), and octanol/water partition coefficient (QP log Po/w=2:0-6:5) were all anticipated. The absence of violations of these rules decreases the likelihood that a molecule will be consumed and is a sign that it is drug-like. No compounds that had broken the guidelines were discovered. The results suggest that the compounds possessed characteristics that could be utilized to create novel medications (Figure 11).

Figure 11: Graphical representation of docking score of ligands (1-10) with 3Q7D.
Prediction of skin permeability: For drugs meant for topical use, this is crucial. The distribution of the computed skin permeability parameter reveals that every component is within the range that 95% of known medications consider being acceptable (QPlogKp).
Bioavailability, lead likeliness and synthetic accessibility: The bioavailability score for Ketoprofens was found to be 0.55, indicating a chance of 55% (greater than 10%) for rat bioavailability. While Ketoprofens showed lead resemblance and a high value of synthetic accessibility, Brenk and PAINS both lacked any alarms. The chemicals were found to be drug-like molecules, lipid and water-soluble, and physiologically active substances (Figure 12).

Figure 12: Compounds’ bioavailability radar assessed using SwissADME.
Ketoprofen’s pharmacokinetic characteristics and toxicity were predicted. Table 3 displays the findings of the pharmacokinetic properties and toxicity analysis. For pharmacokinetic properties, solubility and partition coefficient were computed, while mutagenicity, tumorigenicity, irritative impact, and risk of reproductive damage were projected for toxicity studies. Good pharmacokinetic qualities were demonstrated by the In Silico pharmacokinetic and toxicology study results. The hydrophilicity of both substances would be determined by the log P value, it was predicted. High log P values have been linked to poor absorption or penetration, and they must be less than 5. According to this analysis, all the substances agreed to this limit. The anticipated pharmacokinetic characteristics and toxicity for a compound’s distribution and absorption characteristics are greatly influenced by its water solubility (log S). The estimated log S values for the substances under study fell within the permitted range. The compound’s overall potential as a drug candidate was assessed using the drug score formula. It combines the risk parameters for toxicity, molecular weight, clog P, log S, and drug-likeness, and those are provided in the table. The drug score revealed that ketoprofens adhere to all regulations are not hazardous, and are not carcinogenic. This finding further motivates us to find more innovative inhibitors for the treatment of cancer (Table 4).
| Sr. No. | Descriptor | Compound 1 |
Compound 2 |
Compound 3 |
Compound 4 |
Compound 5 |
Compound 6 |
Compound 7 |
Compound 8 |
Compound 9 |
Compound 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Molecular Weight | 335.44 g/mol | 311.37 g/mol | 325.36 g/mol | 295.38 g/mol | 309.40 g/mol | 343.42 g/mol | 335.44 g/mol | 357.44 g/mol | 297.35 g/mol | 311.33 g/mol |
| 2 | Log p | 3.34 | 2.53 | 2.10 | 2.68 | 3.07 | 3.01 | 3.34 | 2.90 | 2.38 | 1.94 |
| 3 | H-bond acceptor | 2 | 3 | 4 | 2 | 2 | 2 | 2 | 2 | 3 | 4 |
| 4 | H-bond donor | 1 | 2 | 2 | 1 | 0 | 1 | 1 | 1 | 2 | 2 |
| 5 | Surface Area | 46.17 | 66.40 | 83.47 | 46.17 | 37.38 | 46.17 | 46.17 | 46.17 | 66.40 | 83.47 |
| 6 | Molecular Refractivity | 100.63 | 89.48 | 90.9 | 88.32 | 93.22 | 103.19 | 100.63 | 108.0 | 84.67 | 85.28 |
| 7 | No rotatable bonds | 6 | 8 | 8 | 7 | 7 | 7 | 6 | 8 | 7 | 7 |
Table 3: Ketoprofen’s anticipated drug-likeness characteristics.
| Parameter | ADME properties | Compd 1 | Compd 2 | Compd 3 | Compd 4 | Compd 5 | Compd 6 | Compd 7 | Compd 8 | Compd 9 | Compd 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Absorption | Water solubility (log mol/L) | Moderately Soluble | Moderately Soluble | Moderately Soluble | Moderately Soluble | Moderately Soluble | Moderately Soluble | Moderately Soluble | Moderately Soluble | Moderately Soluble | Moderately Soluble |
| Drug Likeness | Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.56 |
| Skin permeability (log Kp) | -5.02 cm/s | -6.40 cm/s | -6.60 cm/s | -5.43 cm/s | -5.50 cm/s | -5.30 cm/s | -5.02 cm/s | -5.05 cm/s | -6.56 cm/s | -6.45 cm/s | |
| P-glycoprotein substrate (Yes/No) | No | Yes | No | Yes | Yes | Yes | No | Yes | No | No | |
| Distribution | BBB permeability (log BB) | Yes | No | No | No | No | Yes | Yes | Yes | Yes | No |
| Metabolism | CYP1A2 inhibitor (Yes /No) | No | Yes | No | Yes | Yes | No | No | Yes | No | No |
| CYP2C19 inhibitor (Yes/No) | Yes | No | No | Yes | Yes | Yes | Yes | Yes | No | No | |
| CYP2C9 inhibitor (Yes/No) | Yes | No | No | No | No | Yes | Yes | Yes | No | No | |
| CYP2D6inhibition (Yes/No) | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | No | |
| CYP3A4 inhibitor (Yes/No) | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | No |
Table 4: ADMET properties of ketoprofen amides.
Conclusion
Ten ketoprofen amide derivatives were examined on the growth of distinct human tumour COX-2 protein after multiple computational observations suggested that different NSAIDs had anticancer potentials. Ketoprofen amides exhibited very weak antiproliferative activity, but the tested amide’s growth inhibitory action amply shows that an amide bond between the substituents is necessary for the much higher cytostatic activity. Furthermore, it was established that the most energetic derivatives are 1 and 6, which exhibit the lowest binding affinities. These derivatives were examined in depth, and tests are being conducted to determine their bioactivity and potential as drugs. These findings, in our opinion, should serve as the starting point for additional study and synthetic optimization of new NSAID amides as prospective prodrugs for anticancer treatment or chemoprevention applications with fewer harmful side effects. The drug score revealed that ketoprofens adhere to all regulations, are not hazardous, and are not carcinogenic. Studies are now being conducted to evaluate the COX selectivity and anti-inflammatory potential of these drugs.
Acknowledgement
We express our gratitude to Department of Chemistry, University of Okara, Pakistan and specially Dr. Ataf Ali Altaf for their support.
Competing Interest
The authors declare no competing interest.
Author’s Contributions
Muhammad Zeeshan: Project design, Project Conceptualization, Docking analysis and wrote the main manuscripts, Sidra Batool: ADMET analysis and ADMET write-up, Muhammad Usman Alvi: Supervision, Resources, review and editing, Zarif Gul: Analysis and proofreading, Maryam Ahmed: Prepared figures, editing and review, Urooj Shahzadi: Resources and Editing, Hassan Abbas: Manuscript proofreading.
Funding
This research work was not funded by any external agency.
Data Availability
All data has been included in the manuscript.
References
- M.J. Thun, S.J. Henley, C. Patrono, Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues, J Natl Cancer Inst, 94(2002):252-266.
- N.R. Sivak-Sears, J.A. Schwartzbaum, R. Miike, M. Moghadassi, M. Wrensch, Case-control study of use of nonsteroidal antiinflammatory drugs and glioblastoma multiforme, Am J Epidemiol, 159(2004):1131-1139.
- S.S. Husain, I.L. Szabo, A.S. Tamawski, NSAID inhibition of GI cancer growth: Clinical implications and molecular mechanisms of action, Am J Gastroenterol, 97(2002):542-553.
- S. Kapitanović, T. Cacev, M. Antica, M. Kralj, G. Cavrić, et al. Effect of indomethacin on E-cadherin and β-catenin expression in HT-29 colon cancer cells, Exp Mol Pathol, 80(2006):91-96.
- G.S. Zhang, D.S. Liu, C.W. Dai, R.J. Li, Antitumor effects of celecoxib on K562 leukemia cells are mediated by cell-cycle arrest, caspase-3 activation, and downregulation of Cox-2 expression and are synergistic with hydroxyurea or imatinib, 81(2006):242-255.
- M. Barbarić, M. Kralj, M. Marjanović, I. Husnjak, K. Pavelić, et al. Synthesis and in vitro antitumor effect of diclofenac and fenoprofen thiolated and nonthiolated polyaspartamide-drug conjugates, Eur J Med Chem, 42(2007):20-29.
- S.C. Sweetman, Martindale: The complete drug reference, Pharmaceutical Press, 2005.
- M. Zovko, B. Zorc, M.J. Takač, B. Metelko, P. Novak, The novel ketoprofenamides: Synthesis and spectroscopic characterization, 76(2003):335-341. [Crossref]
- M. Marjanović, B. Zorc, L. Pejnović, M. Zovko, M. Kralj, Fenoprofen and ketoprofen amides as potential antitumor agents, 69(2007):222-226.
- S.M. Lim, T. Xie, K.D. Westover, S.B. Ficarro, H.S. Tae, et al. Development of small molecules targeting the pseudokinase Her3, Bioorg Med Chem Lett, 25(2015):3382-3389.
- J.P. Hughes, S. Rees, S.B. Kalindjian, K.L. Philpott, Principles of early drug discovery, Br J Pharmacol, 162(2011):1239-1249.
- P. Szymański, M. Markowicz, E. Mikiciuk-Olasik, Adaptation of high-throughput screening in drug discovery-toxicological screening tests, Int J Mol Sci, 13(2011):427-452.
- K. Wichapong, A. Nueangaudom, S. Pianwanit, W. Sippl, S. Kokpol, Identification of potential hit compounds for Dengue virus NS2B/NS3 protease inhibitors by combining virtual screening and binding free energy calculations, Trop Biomed, 30(2013):388-408.
[Google Scholar] [PubMed]
- J. Wu, J. Hu, X. Sun, H. Wang, Y. Huang, et al. In silico study reveals existing drugs as α-glucosidase inhibitors: Structure-based virtual screening validated by experimental investigation, J Mol Struct, 1218(2020):128532.
- C. Wu, Y. Liu, Y. Yang, P. Zhang, W. Zhong, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods, Acta Pharm Sin B, 10(2020):766-788.
- A. Daina, o. Michielin, V. Zoete, SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci Rep, 7(2017):42717.
- K.C. Duggan, D.J. Hermanson, J. Musee, J.J. Prusakiewicz, J.L Scheib, et al. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2, Nat Chem Biol, 7(2011):803-809.
- D.S. BIOVIA, Discovery Studio Visualizer, Dassault Systems: San Diego, 2021, CA.
- M.J Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, et al. Gaussian 16, 2016.
- N.M. O'Boyle, M. Banck, C.A. James, C. Morley, T. Vandermeersch, et al. Open Babel: An open chemical toolbox, J Cheminform, 3(2011):33.
- S. Dallakyan, A.J. Olson, Small-Molecule library screening by docking with PyRx, Methods Mol Biol, 1263(2015):243-250.
- Xu J, M. McPartlon, J. Li, Improved protein structure prediction by deep learning irrespective of co-evolution information, Nature Machine Intelligence, 3(2021):601-609.
Copyright: © 2023 Muhammad Zeeshan, et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.